
Proteomics/Alternative splicing and its impact on protein identification
Alternative splicing: Biological function
[edit | edit source]Alternative splicing is the process in which the primary transcript of a gene is reorganized to produce a different protein than the primary transcript. By manipulating of the exons, the sequence of the amino acids produced from the mRNA is affected, resulting in a different protein sequence, and protein structure.[[1]] This can have a drastic effect on the protein that is produced. Alternative splicing has been observed as a mechanism to produce tissue specific proteins from a signe gene. Depending on the tissue different proteins can be produced in different tissues from a single gene. This process can be thought of a multiplication process that increases the possible proteins that are produced from a single gene.

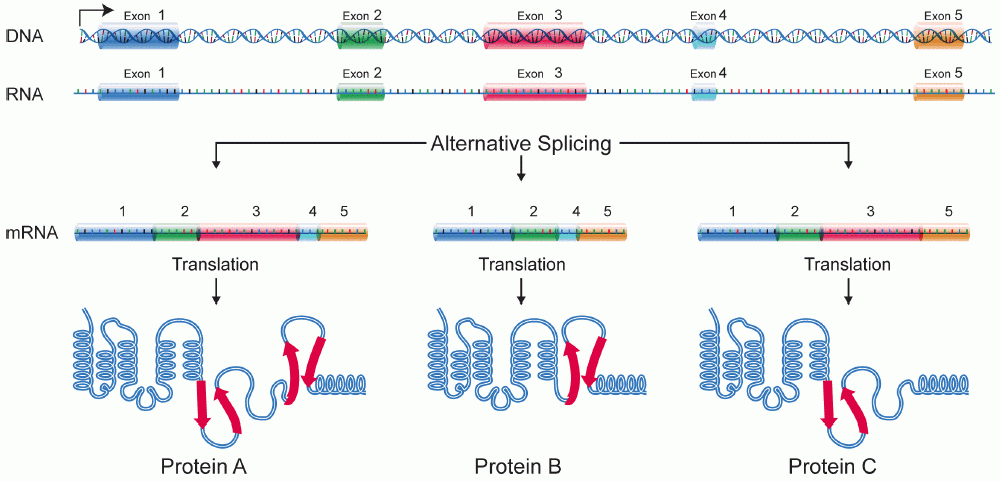{kind=link}
Alternative Splicing is a major source of protein diversity in living organisms. It has been estimated that at least 30% of all genes in the human genome are alternatively spliced and this number continues to expand. It was originally thought that the number of alternatively spliced genes accounted for only 5% of proteins in humans. With the unveiling of the human genome it was revealed that the human genome contains less than 30,000 genes[[2]]. This could potentially account for the huge gap between the number of genes and the proteome.
It has been suggested that alternative splicing is the source of higher level complexity in eukaryotes[[3]]. This idea is based on the thought that more complex organisms will alternatively splice their genes more often to obtain more possible mRNA sequences. However evidence shows that the level of alternative splicing between different complexities of organisms is not significant. Providing evidence of the contrary. This study was done using ESTs (expressed sequence tags *link to EST page*). As more EST studies are done, it has become apparent that there is a greater number of alternatively spliced genes than previously thought. ESTs were compared to mRNA sequences using BLAST.
Examples of Alternative Splicing
[edit | edit source]Alternative splicing has been implicated in several diseases. An example of a disease that plays a role in alternative splicing is Rett syndrome. This disease is found primarily in girls and is characterized by problems in forming connections between neurons, or synapses[[4]]. It is believed that the gene MECP2 produces a protein that regulates alternative splicing of some proteins. When this gene is disrupted, transcripts of other genes that would normally be spliced by MECP2 are not spliced, leading to Rett the phenotype of the syndrome.
Splicing Mechanism
[edit | edit source]Splicing occurs through the mechanism of the Spliceosome. The spliceosome consists of a number of proteins and snRNA components. snRNAs U1, U2, U3, U4, U5 and U6[[5]]. These snRNAs recognize the splice sites and then recruit other proteins that connect the splice sites. These splice sites are then brought together through the interaction of these proteins which form the spliceosome. Once the spliceosome has formed, the sites are cleaved to bring the correct exons (or introns) together.

{kind=link}
There are 4 common types of alternative splicing, they are as follows:
- Alternative promoter selection: A different promoter is used for different splice variants. This results in a different start of the mRNA transcript.
- Alternative selection of cleavage/polyadenylation sites: Different exons are spliced based on recognition of different cleavage or polyadenylation sites, entire exons can be skipped. Results in a different exon at the 3’ end of the transcript.
- Intron retaining: Introns are used as coding regions. A sequence that is normally considered an Intron is retained in the final transcript that serves as a template for translation.
- Exon cassette: Entire exons can be skipped in the middle of the protein, resulting in a different transcript

{kind=link}
The Growing Need for Protein Identification:
[edit | edit source]Proteins are the basic unit of structure and function within living systems. Consequently the field of proteomics has taken on added significance in the modern study of biology. The genomic revolution, which culminated in the sequencing of many genomes, has generated a tremendous amount of data. The field of proteomics [[6]] has unfortunately been lagging behind, resulting in a disconnect between genomic information and observable phenotypes. Originally, proteins and protein dependent pathways were studied individually. Recently an emphasis on systems biology has lead to changes in this methodology. Entire cells are being characterized with high throughput techniques.

{kind=link}
Mass spectroscopy has become the golden standard of protein identification ([[7]]) [[8]]. Briefly, proteins are broken down into peptides, suspended into a gas phase by one of a various number of methods, ionized, and sent through a detector which can determine the mass to charge ratio of various peptides. Mass spectroscopy can be easily automated and combined with other forms of protein separation, making it an ideal candidate for high throughput analysis. In addition, thousands of peptides can be identified from a single source at one time, making this technique much more applicable to systems biology then older techniques such as Edman degradation. Mass spectroscopy can also be used to identify single proteins of interest that have been isolated using other techniques, such as chromatography.

{kind=link}
Splicing and Protein Identification
[edit | edit source]A major drawback of mass spec analysis, and even identification by Edman degredation, is that proteins must be digested into peptides before identification. Often for use with shotgun sequencing [[9]], proteins are digested before any form of isolation process is done. In order to confirm identifications protein databases are searched in order to match unique peptides to entire proteins. This process is made more complicated by the immense level of sequence homology shared by peptides produced by alternative splicing. [[10]]. These proteins, although sharing similar primary structures may have very different or even antagonist functions, making their identification critical from a biological perspective. What is more, the level of alternative splicing that occurs is not well characterized so it is not even well known which proteins cannot be unambiguously identified. These problems will likely remain until alternative splicing is well documented or can be predicted efficiently computationally.
Efforts to analyze alternative splicing:
[edit | edit source]In vivo analysis is currently the most accurate way to identify alternative splicing, either at the transcript, or in some cases at the protein level. Numerous databases exist that document proteins known to undergo alternative splicing, including the Alternative Splicing Database [[11]]. and the Alternative Splicing and Transcript Diversity database [[12]]. Well these are good references, as discussed above, the estimates of even the amount of alternative splicing present in eukaryotic organisms vary dramatically. There is therefore, very little information on how complete these databases are.
Steps have also been taken to computationally predict alternative splicing [[13]]. Generally these algorithms combine gene finding approaches with experimental data. Splice sites are recognized and rated in efficiency based on consensus sequences. Sequences are then matched to known expressed sequence tags to make predictions. Tools such as BLAT, Spidey, and SIM4 can be adapted for these processes. Most modern computational tools encounter difficulty in comparing genomic data to sequences as small and variable as splice sites. False positives and negatives are fairly common [[14]]). New approaches are still being developed.
References
[edit | edit source]1. Möröy et al. The impact of alternative splicing in vivo: Mouse models show the way.
http://www.rnajournal.org/cgi/content/full/13/8/1155 obtained in April, 2008
2. Alternative RNA Splicing. ExonHit Therapeutics. http://www.exonhit.com/index.php?page=59. obtained in April, 2008 3. Brett, D et al. Alternative splicing and genome complexity.
http://www.nature.com/ng/journal/v30/n1/abs/ng803.html;jsessionid=BF0AED8347574D063F5E347EC693AE83 obtained in April, 2008
4. Rett Syndrome Fact Sheet. National Institute of Neurological Disorders and Stroke.
http://www.ninds.nih.gov/disorders/rett/detail_rett.htm#109713277 obtained in April, 2008
5. Cáceres, J et al. Alternative splicing: multiple control mechanisms and involvement in human disease. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TCY-45FYM7X-F&_user=47004&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000005018&_version=1&_urlVersion=0&_userid=47004&md5=eea15989e03f8b963bdc33384a4ef93b. Obtained in April, 2008 6. Obtained from wikipedia. http://en.wikipedia.org/wiki/Image:AlternativeSplicing.png. Obtained in April, 2008 7. Obtained from wikipedia. http://en.wikipedia.org/wiki/Proteomics. Obtained in April, 2008 8.